Membrane Pharmacy Structure DynamicsResearch group : Priv.Doz. Dr. Thomas NawrothProtein Purification |
Membrane Pharmacy Structure DynamicsResearch group : Priv.Doz. Dr. Thomas NawrothProtein Purification |
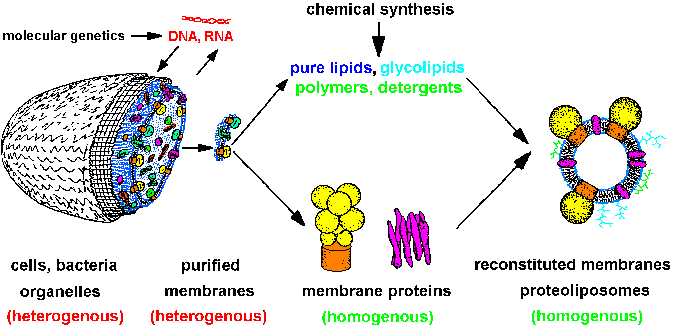 |
| Fig.1: The strategy of structure and membrane research leads from heterogenous cells over isolated homogenous systems (proteins, lipids, membranes, nucleic acids) to reconstituted model membranes and protein-detergent complexes (after T20). Molecular genetics supplies cells with specific proteins, chemical synthesis gives us lipids, polymers and compounds for optical system manipulation, e.g. caged-compounds. The molecular homogenous systems are suitable for structure investigation (SAXS, SANS, ASAXS, X-ray diffraction, EXAFS / XANES, especially with time resolution). |
The isolation and purification of the membrane proteins requires the
solubilization of the protein by replacement of the lipids by detergents
(both are amphiphiles). As shown in figure2, the native structure and thus
the biological function is only retained if weak, non-denaturating detergents
of highest chemical quality are used (containing no peroxides or long chain
impurities). The non-denaturating detergents do not penetrate into the
protein and have at least one of the following properties: i) large area
of the hydrophobic domain, and ii) low charge density in the hydrophilic
head. This is the case for example in the detergents CHAPS, CHAPSO, TDOC,
DOC, Digitonin, Deoxy-BigChap, Laurylmaltoside, Octylglucoside, Triton-X100
(has to be peroxide-free !).
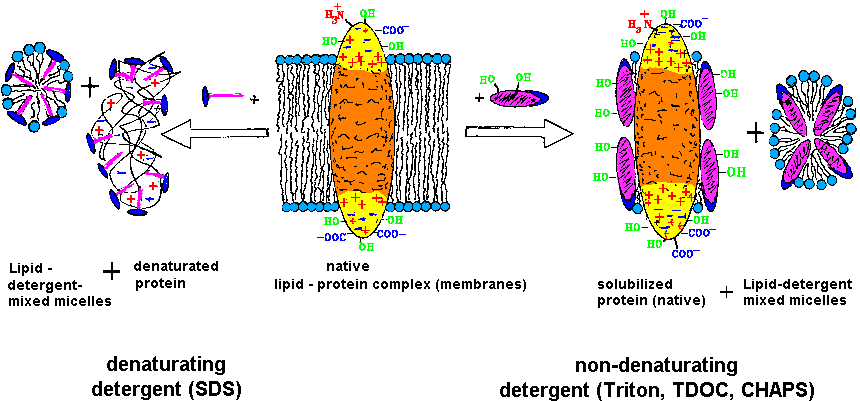 |
| Fig.2:The purification and investigation of membrane proteins requires the retainment of the function competent structure (native), which is obtained by solubilzation with weak, non-denaturating detergents [T20]. |
The choice of the detergent and conditions for the protein solubilization is the key for the isolation: i) The replacement of the membrane lipid by the detergent must not demage the protein structure and function; ii) the solubilization should be selective, i.e. in the ideal case the protein of interest should be solubilized only together with a few other proteins, which can then be removed by chromatography; and iii) the protein must not be demaged during isolation, e.g. by protease action, partial unfolding or oxidation, which might occur by detergent impurities. As shown in table1 and T20, the solubilization can be done following three methods: direct solubilization, fractionated detergent solubilization, and fractionated detergent exchange solubilization.
Table1: Methods of membrane protein solubilization by detergents.
| Solubilization method | procedure |
| direct solubilization | membrane extraction with one detergent at a given concentration, the resdue after cenrifugation is removed |
| fractionated detergent solubilization | membrane extraction with one detergent in two steps, the first extract and the residue is removed |
| fractionated detergent exchange solubilization | membrane pre-extraction with one detergent - extract is removed;
then fractionated extract with another detergent - resiue removed |
In living cells the constituents are subject of continous degradation
and novel biosynthesis. The degradation is maintained by cellular proteases,
which are down regulated in vivo. During disrupture of the cells, membarne
preparation, deteregent solubilization and protein purification that regulation
is lost. Thus several proteases may degrade the proteins of interest, which
may lead to artefacts. Those protease artefacts can be avoided by i) cooling
(ice); ii) rapid passage through the process steps from cells to the first
chromatographic purification; and iii) inhibition of proteases by inhibitors
during all steps of protein purification. A variety of intracellular proteases
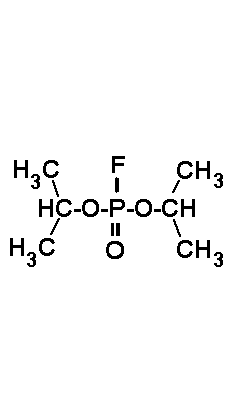
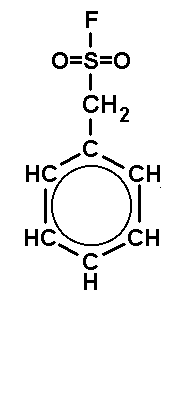
can be inhibited by the acid halogenides shown in Fig.3, which act on active
serine residues. Those protease inhibitors are useful also during structure
investigation, e.g. protein crystallization: They inhibit proteases and
avoid microbiol problems as well. Note that the lifetime of those acid
halides is temperature and pH dependent (at 20°C, pH7 about 1-2
days). Unfortunately the most powerful inhibitors are extremely toxic (xenobiotics),
namely DFP (toxic dosis for man: 1 µl). Thus they should handeled
with care by experienced persons only (chemistry security training required
!).
 |
 |
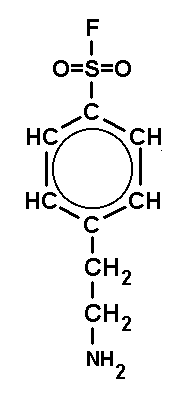 |
| DFP (DiisopropyFluoroPhosphate) is a volatile toxin. The working concentraion is 0.1 mM. | PMSF (PhenylMethylSulfonylFluoride) is a solid. The working concentraion is 0.1 mM. | PefaBloc (AEBSF) is a solid well soluble in water. The working concentraion is 0.5 - 5 mM. |
After the selective solubilization, the protein content of a preparation has reduced to a few %, if a proper procedure has been applied. The protein can then be subjected to chromatographic purification, which may enrich the protein of interest by a further factor of 10 - 100. For structural biology and molecular biochemistry a product of >95 purity (99% for crystallography and time resolved studies) with fully retained function is required.
Table2: Protein content during steps of a typical preparation for structural biology: ATP-synthase from Micrococcus luteus.
| step / procuct | 250g cells (wet weight) | purified membranes (PMV) | fractionated detergent exchange solubilisate | 1st ion exchange chromatography IEC | 2nd ion exchange chromatography IEC | GPC: gel permeation with detergent exchage |
| protein content | 40g | 8 g | 3 g | 200 mg | 100 mg | 60 mg |
| purity | 0,15% | 3% | 10% | 80% | 98% | >99% |
The first column of the chromatographic purification has to be a material capable of "heavy duty": i) it must be capableof handling large protein amounts and volume (several grams); ii) it has to be insensitive to impurities, e.g. lipids and easy to regenerate many times; and iii) is has to be stable to microbiol attack. For large scale preparation for structural biology studies we use in several preparations, e.g. ATP-synthase and F1ATPase a large tentaculus ion exchanger (Fractogel EMD650m-DEAE, Merch, Darmstadt) HPLC column of 60 g protein binding capacity (biotechnology scale). Then a column of higher separation power follows: for ATP-synthase Fractogel EMD650s-DMAE (HPLC); for Cytochrome-c oxidase or Quinol-oxidase: affinity chromatography colums (Lysine-Agarose and cytochrome-c-Agarose). In most preparations of membrane proteins the last purification step has to combinded with a detergent exchange to a detergent suitable for structure research. This can be done for large memrane proteins by gel permeation chromatography (GPC) using Superose6 (Pharmacia). The final product is concentrated by ultrafiltration and stored in liquid nitrogen. An addition of 10% glycerol during purification and freezing is useful (protein stabilizing). Furthermore this prevents the protein from radiation demage during the investigation at high-flux synchrotrons, e.g. by time resolved methods. Some protein purifications developed by the MPSD group are presented in table3. Preparation protocols are available on demand (email).
Table3: Some protein preparations of the MPSD group (overview in T20).
| Protein | organism / strain | preparation type | yield from cells | references, comments |
| ATP-synthase | Micrococcus luteus | small scale | 10 mg from 60 g | |
| ATP-synthase | Micrococcus luteus | biotechnological scale | 70 mg from 250 g | for TR-studies and neutron scattering / liposomes |
| F1ATPase | Micrococcus luteus | small scale | 25 mg from 60 g | |
| F1ATPase | Micrococcus luteus | biotechnological scale | 200 mg from 250 g | for TR-studies and crystallization |
| F1ATPase | Micrococcus species | small scale | 20 mg from 60 g | |
| F1ATPase | Escherichia coli | small scale | 10 mg from 200 g | |
| F1ATPase | Rhodospirillum rubrum | small scale | 5 mg from 60 g | chloroform method |
| ATP-synthase | Rhodospirillum rubrum | small scale | 10 mg from 60 g | |
| ATP-synthase | beef heart mitochondria | small scale | 20 mg from 1 beef heart | |
| Cytochrome-c oxidase COX | Micrococcus luteus | small scale | 5 mg from 60 g | |
| Quinol oxidase QOX | Micrococcus luteus | small scale | 10 mg from 60 g | |
| Cytochrome-bc1 complex | Rhodospirillum rubrum | small scale | 5 mg from 60 g | |
| Quinol oxidase QOX | Rhodospirillum rubrum | small scale | 5 mg from 60 g | |
| Bacteriorhodopsin purple membrane PM | Halobacterium salinarium | large scale | 200 mg from 100g | |
| monomeric Bacteriorhodopsin mBR | Halobacterium salinarium | small scale | 30 mg from 50 mg PM | |
The methods and technology of membrane protein purification is subject
of the lectures "Methods of membrane biochemistry".
The methods for structure research of membranes and membrane proteins in
solution are subjects of the lectures
"Biophysical chemistry of membranes and membrane proteins". You can learn
the technology of membrane protein purification and some techniques for
biophysical chararacterization by participation in our practical
course "Advanced practicum in membrane biochemistry". The analytical
techniques are subject of the Bioanalytics lecture.