Membrane Pharmacy Structure DynamicsResearch group : Priv.Doz. Dr. Thomas NawrothATP-synthase |
Membrane Pharmacy Structure DynamicsResearch group : Priv.Doz. Dr. Thomas NawrothATP-synthase |
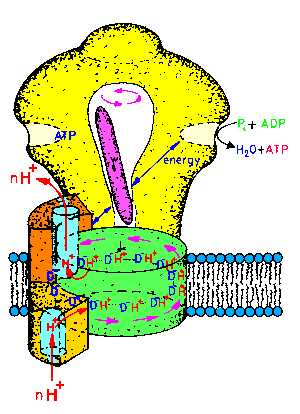 |
| Fig.1: ATP-synthase is an intrinsic membrane protein, consisting of two subcomplexes of various protein-subunits: The membrane integral Fo-part is a proton pump, whereas the hollow headpiece, called F1ATPase bears the 3 catalytic centers. The reversible system converts energy of the proton translocation to the synthesis or hydrolysis of ATP (30 kJ/mole ATP at pH7). The intramolecular energy transfer is maintained by a series of molecular motions. The rotation of the central gamma-subunit (pink) connects the events inside the F1-head with those in the Fo-complex (picture from T20, structure of the Fo-part as suggested by W. Junge). |
ATP-synthase is a large membrane protein,
which plays a key role in the energy metabolism of all known organisms,
i.e. it is the terminal protein in a network or chain of membrane proteins
of the bioenergetic system. The enzyme couples
the vectorial proton transport accross a membrane with the synthesis or
cleavage of the energy rich compound ATP (AdenosineTriPhosphate) in a reversible
reaction. As depicted in Fig.1, ATP-synthase consists of two subcomplexes
of several (>22) protein subunits, named Fo-part (membrane integral
base-piece) and F1ATPase (head). The two active areas of the
proton pump in Fo and of the chemical catalysis in F1
are apart about 10nm, but energetically coupled. This distance is much
too large for any chemical coupling. Thus it was suggested already in the
early 80's that the coupling occurs in a structure-mechanical way, i.e.
by conformational coupling. Biochemistry yielded indirect evidence for
this suggestion by estimation of the binding constants of nucleotides (ATP,
ADP) to catalytic and non-catalytic nucleotide sites. As depicted in Fig.2
the F1ATPase-head contains 3 catalytic nucleotide sites (easyly
exchangable) and three non-catalytic sites (under steady-state conditions
non-exchangable nucleotides). Paul Boyer concluded from the fact that the
nucleotide binding sites occured variable in strength and accessibility
during the reaction (open, loose, tight states in Fig.2), observed in an
elegant way by time resolved isotope exchange experiments, that each active
center undergoes a sequence of structural interconversions while it is
cooperatively coupled to one or two other active centers, which are in
the opposite structural state and reaction. 1997 he got the Nobel award
for this "binding change mechanism" together with J.E.Walker, who had solved
the structure of a resting inhibited modification of the F1ATPase
by static X-ray crystallography ([1994] Nature 370, 621-628). Nevertheless
the reaction mechanism remained unclear, especially the energetic coupling
between the three alpha-beta heterodimers inside the F1ATPase
and the coupling between Fo- and F1-part. The reason
for this was probably the lack of time resolution in most experiments.
The crystallography showed a cleft between the central gamma-subunit
and the hollow F1ATPase-head, which could allow a motion of
this central axis, possibly a rotation (the subunit is shown in pink in
Fig.1). 1997 this suggestion of Walker was proved by Noji et al. (Nature
386, 299-302) by video-microscopy of immobilized F1ATPase with
a fluorescence labeled gamma-subunit. 1998 Oster et al. presented a theoretical
calculation of molecular motions in F1ATPase obtained with a
modified RASMOL program run based on Walker's data. These symmetric dislocations
of three cooperative domains (out, to the side, in) extrapolated simply
the three different states of the three beta-subunits in the static F1ATPase-structure,
in a kind of morphing. As symmetric changes do not change the centre of
gravity, they should not yield any measurable effect in a spatial avaraging
method of structural biology, e.g. X-ray small angle scattering (SAXS).
In contrast the time resolved experiments (TR-SAXS)
of the MPSD group started at DESY-HASYLAB
(F14,
T20,
T14)
and continued at ELETTRA and
ESRF
(submitted) showed the transient size changes and subunit structure rearrangements
of the working enzyme presented below.
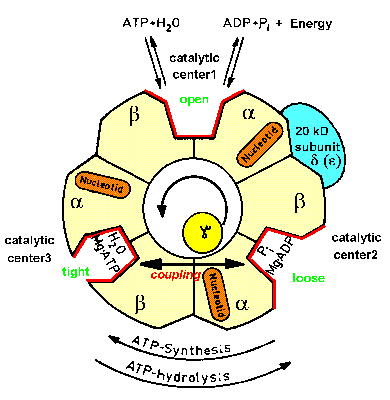 |
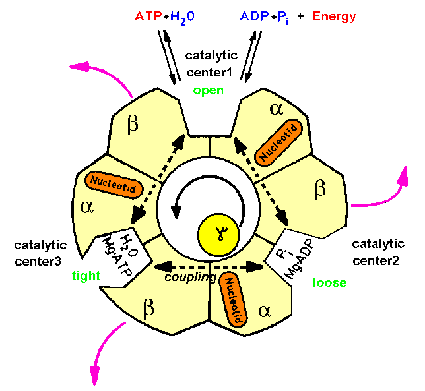
| Fig.2: Structure scheme of F1ATPase. The cut through the hollow F1ATPase head shows three pairs of subunits (alpha, beta heterodimers) and the rotating gamma subunit in the center. Each alpha, beta-pair carries one catalytic center (in the cleft) and an additional burried noncatalytic nuceotide center (orange). Only one subunit pair is associated with the delta-subunit (epsilon in Micrococcus luteus), which is part of the proposed "second stalk" structre (from T20, modified). |
Since the 60's ATP-synthase was assumed to contain a single connective
domain consisting of gamma and small subunits, due to electron microscopy
investigations. In the late 90's it came out that obviously a second connection
exists, consisting of the upper part of the Fo-subunits b,b'
and the F1-subunit delta, which is the epsilon subunit in the
aerobic bacterium Micrococcus luteus used by the MPSD group (delta
- epsilon exchange in Micrococcus, see F8,
F18,
F20
and regulation of F1ATPase). Thus
an inherent asymmetry should exist in the F1ATPase (see Fig.2).
Several groups unsuccessfully searched for this asymmetry, e.g. by labeling
and chemical crosslinking (R. Cross et al.), but the results were ambigous.
Just the time resolved X-ray scattering experiments presented in the structural
film of working ATP-Synthase indicated a kinetic asymmetry in the
working enzyme, which is an evidence of a supercycle of three structural
subcycles in the ATP hydrolysis reaction.
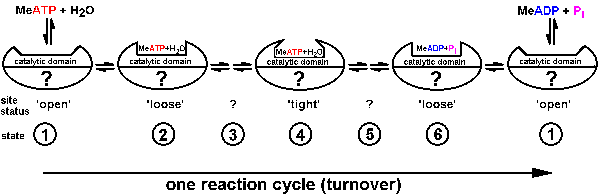
| Fig.3: According to Boyer et al. (alternating site mechanism) each of the reaction centers inside ATP-synthase / F1ATPase undergoes a sequence of status changes during the ATP reaction cycle. While the binding of the nucleotide (ATP/ADP changes from loose to tight and again loose, the chemical accessibility of the site (indirect estimate of the structure) varies from open to closed and again open. The structure of the other sites is unknown (?), while beeing highly cooperative in the multisite-reaction (from T20). |
 |
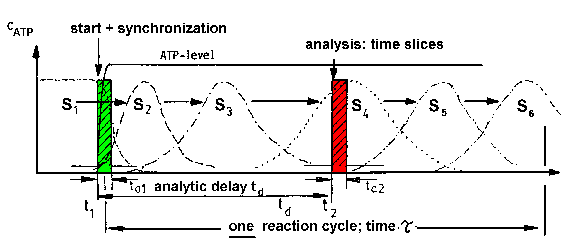 |
| Fig.4: The investigation of the transiently occuring states of the working protein requires the activation, e.g. by substrate (ATP) binding, and analysis with time resolved methods. The time required for the activation (ta1) and analysis (ta2) has to be smaller as the duration of one reaction cycle Tau (= 1/kcat) (from T20). |
The observation of events during the reaction cycle of a working enzyme
(catalytic protein) requires time resolved experiments.
As presented in Fig.3 for the assumed binding change mechanism of F1ATPase
/ ATP-synthase according to Boyer, a catalytic domain of each molecule
of the enzyme passes sequentially through a series of states ((1) to (6),
which differ in structure and chemical properties (binding "constants"):
The substrate (metal-ATP for F1ATPase) binds to an open conformation
(1) of the empty protein-domain; then the ATP is bound loosely to a conformation
(2); this converts over a closed conformation of high energy (3) to a tight
modification of low energy content, from which the ATP cannot escape; a
transition over an intermediate ADP state (5) yields then the conformation
(6), where the product (ADP) is loosely bound (exchangable), which dissociates
to product to the environment (solution); and the final transition recreates
the open conformation of the empty domain (1). In the ATP-synthesis reaction
the sequence happens in the opposite direction. The status of each catalytic
domains is linked to that of one or two others in a cooperative way, indicated
by the quotation mark (?) in Fig.3. The way of coupling depends on the
nucleotide concentration, which switches the enzyme over between at least
three operational modes: single site, dual site and multi (tri) site catalysis.
Due to biochemical experiments, the energy input is required not for the
formation of ATP+H2O from ADP+phoshate, but for the structural conversion
of tightly bound (not exchangable) ATP to loosely bound (exchangable) ATP.
Thus a structural interconversion of the protein and not a chemical reaction
is the key of the process.
As depicted in Fig.4, the series of conversions can in principle investigated
by time resolved methods of structural biology
and biochemical analysis if: i) the enzyme is activated in a short time
ta1 and ii) the signal (structure, product) is analyzed in a
short time ta2; both intervals have to be much shorter (>10x)
as compared with the duration tau of the enzymatic reaction (= 1/kcat).
If in a macroscopic sample, i.e. a protein solution or a crystal, the majority
of the molecules can be activated
in parallel, the system behaves like a single molecule for a limted
time, at least for the reaction cycle time tau. In this case the sample
is synchronized and shows the state densities S1 to S6,
which correspond to the molecular states (conformations) 1 to 6. If the
analysis time is smaller than the life time of the populations beeing
in these conformations, the structures can investigated with the methods
of structural biology, e.g. time resolved small
angle scattering of working proteins. This requires a high flux synchrotron,
especially if the reaction cycle has to be investigated in single shot
experiments (most common case).
Experimental problems and solutions
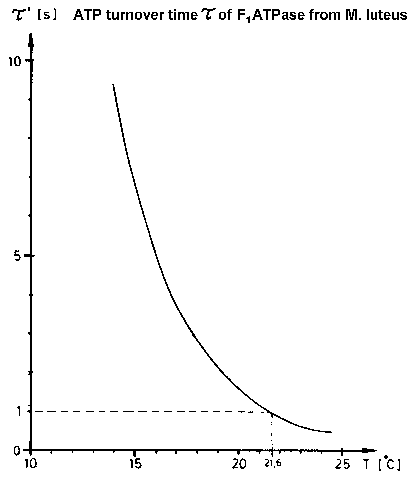 |
| Fig.5: The speed of molecular motions in working F1ATPase can be reduced by cooling (or D2O-inhibition). Due to the remarkable high activation energy for the ATP-hydrolysis of the low temperature modification of complete F1ATPase from Micrococcus luteus F1L (see page: regulation of F1ATPase), the duration of one enzymatic ATP cleavage can be lowered by 3 orders, e.g. from 4 ms at +37°C to 20 s at +13°C !. (from T20) |
At 37°C the enzymatic ATP hydrolysis takes 4ms with F1ATPase
from Micrococcus luteus. This is too fast for current TR-SAXS experiments
at high flux synchrotons with CCD-detectors (read-out dead time (gap) >
100 ms). As shown in Fig.5 the reaction can be slowed down to 20 s at +13°C
by moderate cooling because of the extraordinaryly high activation energy
of the enzymatic reaction of the low temperature modification of the complete
emzyme F1L (see page: regulation
of F1ATPase). This "cool trick" works with proteins,
which show molecular motions during the reactions (motor proteins, allosteric
proteins); in contrast the temperature effect on proteins which work by
intramolecular electron transfer is much smaller.
The time resolved activation of the majority of molecules in the protein
solution or crystal, i.e. the synchronization
of the sample, requires the conversion of the free enzyme molecules
E into the enzyme-substrate complex ES: According to the commonly accepted
Michaelis-Menten theory of enzyme action
E + S <> ES <> (ES*) <> (EP*) <> EP <> E +
P (equ.1)
the reaction E + S <> ES is rate limiting at low substrate concentration,
while ES <> ES* is rate limiting under substrate saturation conditions
(P is the product, (ES*) is the activated transition state of short life
time). This synchronization can be acchieved by several strategies: 1)
The system can be activated by a concentration jump of the substrate (ATP)
by rapid mixing of enzyme and substrate stock solutions with a stopped-flow
device, as shown below; 2) The system can be activated by a concentration
jump of the substrate (ATP) by flash photolysis of a protected substrate-derivative,
e.g. caged-ATP; 3) The system can be activated by conversion
of an inactive protein modification into an active enzyme, e.g. byflash-photolysis
/ dissociation of a protein inhibitor, e.g. carbon monoxide CO from Cytochrome-oxidase
or Myoglobin; 4) the complete system, an inactive mixture of protein
and reactand, can be activated by a jump event, e.g. by a temperature
jump of a cold-inactivated mixture. With F1ATPase from Micrococcus luteus
the strategies 1, 2 and 4 were successfully tested in the MPSD group (see
T20).
Below the results with the simplest method are presented, the rapid mixing
with a stopped-flow device.
During structure investigation with a high flux synchrotron (or later
a FEL) the specific problems presented in Table1 occur. In the last 4 years
we found the solutions in the table, which enabled us to use the full flux
of the most brilliant monochromatic X-ray beamline existing, the ID2A beamline
at ESRF, for our investigation of working proteins (F1ATPase,
ATP-Synthase (and in collaboration with H. Heumanns group, MPI Martinsried,
the caperones GroEL/ES). After first experiments and several improvements
(1997/1998) the state-of-the-art experiments (1999/2000) in cooperation
with T. Narayanan (ESRF) yielded a complete scattering profile at a flux
of 2x1013 ph/s at sample (0.3x0.8mm focus, 12.5 keV photons)
in 150 ms (i.e. one frame of a structural film of 110 images). This is
about the same photon flux, that will be obtained during a microbunch of
the planed pulsed free electron laser FEL at DESY, Hamburg, in 10 years
(but the film will be 100,000 times faster). The stability of F1ATPase
from
Micrococcus luteus in an X-ray experiment with the full ID2A-flux
is shown in Fig.6. The time resolved estimation of the radius of gyration,
which is an estimate of the averaged molecule size, indicates that this
enzyme survives the beam (radiation demage) in presence of a radiacal scavenger
and in degased solution (oxygen -> O2minus*-radical !), which
was cooled by a helium jet in our novel sample environments XBox2 and XBox4
(see S30,
R7)
for >30 s before fragmentation of a small sub-population. This is the time
window for structureal biology with the working protein. The increase at
the end (130 s) indicates the formation of a gas-bubble. The radiation
tolerance is different for several proteins and followed the series:
F1ATPase / ATP-synthase (Micrococcus luteus) > GroEL/ES (E. coli) > Hemocyanin (Eurypelma californicum) (about 50:5:1)
Table1: Problems in the investigation of molecular motions at high flux synchrotrons and some solutions.
|
|
|
|
| sample heating by beam absorption | helium jet cooling (see: sample environment in S30),
defocussing of beam (e.g. at ELETTRA-SAXS) |
defocussing and re-focussing required at XFEL, pulsed bunch multiplexing |
| gas bubble formation under irradiation | degasing of samples (15 Torr, 1h) | Helium degasing of samples (as usual for HPLC solvents) |
| radiation demage | use of 2D-detectors (catch any scattered photon),
radical scavengers (10% glycerol, trehalose), continous replacement of sample by slow pump (see S30), or sample cell motion (motor, piezo drive) |
radical scavengers, defocussing and re-focussing, and sample cell motion by a fast piezo drive (XFEL) |
| time resolved data estimation, interuptions | 2D-gas detectors currently are limited to 5x1010 ph/s at
sample (slow kinetics >> 10s),
CCD cameras (e.g. frelon-XRII) withstand high flux (>1014 ph/s at sample) but have reading gaps (100ms), 3rd generation synchrotron yield enough photon for a time resolution > 1ms in single shot experiments |
integrative detectors (at an XFEL all scattered photons in a microbunch
(107-108) occur in a few fs),
image plate with an analyzer crystal on a piezo-twister, the time range 1ns - 1ms in single shot experiments is accessible with XFEL |
.
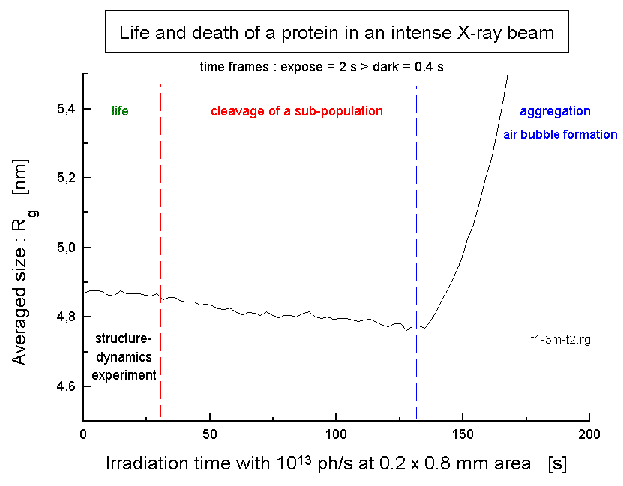 |
| Fig.6: The F1ATPase from Micrococcus luteus (10 g/l, pH7.5; without substrate ATP) withstands the full flux at the high brillance beamline at ESRF (ID02A) of 2x1013 ph/s (in 0.2 x 0.8 mm2) in a degased soltution containing 10% (v/v) glycerol as radical scavenger. The increase after 130 s indicates the formation of a gas bubble. The structural film of the working protein is obtained as a sequence of frames exposed for 300 to 150ms, which is the equivalent of the expected flux during one microbunch at the planed XFEL. |
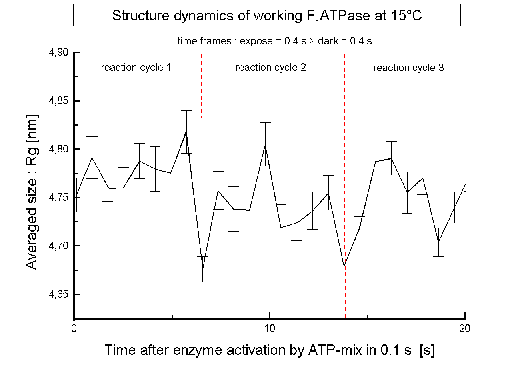 |
| Fig.7: After activation and synchronization of the ATP-reaction cycle by a concentration jump of the substrate CaATP (c = 1 mM = 6 Km) the averaged size of F1ATPase (Rg) changes trasiently. The dashed lines indicate the time required for one ATP-cleavage/protein at the experiment temperature at 15°C (7s). |
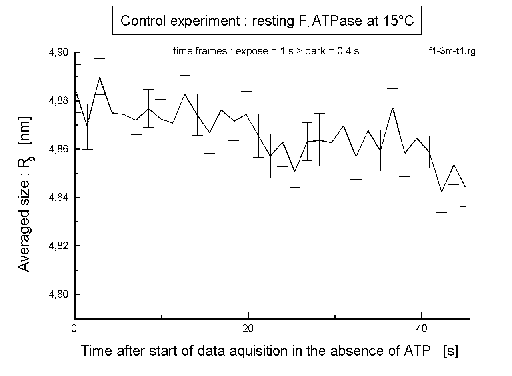 |
| Fig.8: In the control experiment (with no ATP added) the averaged size of F1ATPase (Rg) doesn't depend on time. |
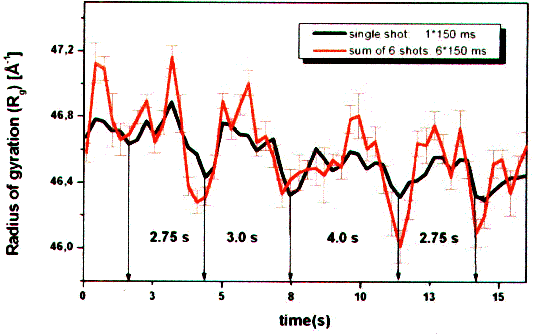 |
| Fig. 9: The estimation of the average molecule size according to the radius of gyration Rg over several reaction cycles at 20°C shows that the third of three ATP-hydrolysis cycles takes about 1/3 more time (4 s versus 3 s). This is an evidence for a supercycle of three subcycles and of a dynamic asymmetry in working F1ATPase (from T23). |
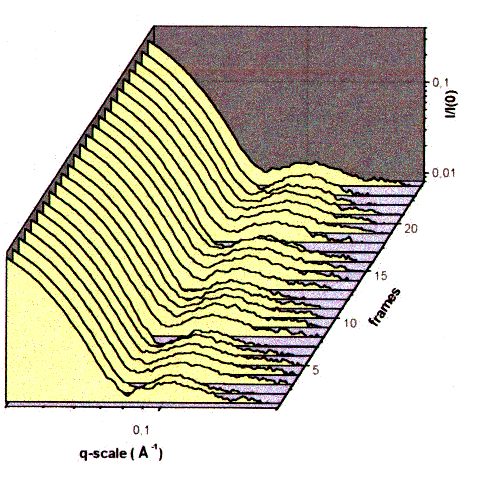 |
| Fig.10: The side maxima in the time resolved small angle scattering of working F1ATPase at 20°C indicate subunit motions in the F1ATPase head directly. The subunit distance ds accross the F1-head corresponds to ds = 2 pi / qs., where qs is the position of the side maximum (momentum transfer). During the frame sequence time of 600 ms the images were taken in the first 300 ms (from T23). |
Experimental conditions:
- F1ATPase: c= 5 mg/ml + 1 mM ATP + 5 mM CaCl2, pH8, 10%
glycerol (protective radical scavenger), T=12-20°C
- The enzyme is at 1 mM ATP saturated for multisite catalysis (Km
= 150 µm with CaATP 5:1).
- The sample is cooled by a helium jet
during the experiment (avoids beam heating; at ESRF-ID2A 50% of the beam
power (25 mW at 2x1013ph/s, 12 keV) is absorbed).
- The speed of the reaction is slowed down
by cooling from 4 ms (37°C) to 5-20 s using of the extraordinary high
activation energy of the catalysis by the low temperature modification
of the inhibitor protein associated F1ATPase F1L
from Micrococcus luteus (see: Regulation
ATP-synthase).
 |
| Fig.11: The changes in the average size (Rg) and the dislocations of the side maxima (subunit distances) can be interpreted by molecular motions of domains (subunit-parts) inside the hollow F1ATPase complex (from T20, modified). |
- The large structural changes of working F1ATPase
and ATP-synthase are due to subunit or domain movements.
- Molecular modelling showed that the motion can not be explained
by the rotation of the central gamma-subunit. Obviously the large subunits
(alpha, beta) or significant parts of them (lower domains) are displaced
radially.
- The molecular motion is a sudden move rather than a diffusive
creeping.
- The kinetics (fast events after a long time) can be explained
by an avalance model, i.e.a sudden transition from a metastable state.
- The long time observation yields an evidence of a supercycle of 3
subcycles (ATP reactions) and a kinetic asymmetry.
- The results suggest a hierarchy of molecular motions in ATP-synthase:
|
Fig.12:The molecular motions in working ATP-synthase occur in a hierarchical manner: During the ATP-synthesis the sequence passes from left to right, whereas upon ATP hydrolysis the events occur from right to left. In F1ATPase the sequence stops at the gamma-subunit rotation. |
Current work:
- molecular modeling with an improved version of our FVM cube method
(F10), capable of simulating molecular
motions.
- study of the short life time conformations
(expanded, shrinked) and transitions by stopped-flow
/ temperature-jump double experiments (cold trap) with our new sample
environment SBox4, associated with 3 thermostats (see also our technology
page).