Membrane Pharmacy Structure DynamicsResearch group : Priv.Doz. Dr. Thomas NawrothTime Resolved Methods (TR-Methods) |
Membrane Pharmacy Structure DynamicsResearch group : Priv.Doz. Dr. Thomas NawrothTime Resolved Methods (TR-Methods) |
In any of these cases the system beeing investigated undergoes a stepwise
transition from a start state (S ; Z1) to a product state (P ; Z6):
|
Start state (Z1) (S) > intermediate (Z2) > intermediate (Z3) > intermediate (Z4) > intermediate (Z5) > Product state (Z6) (P) |
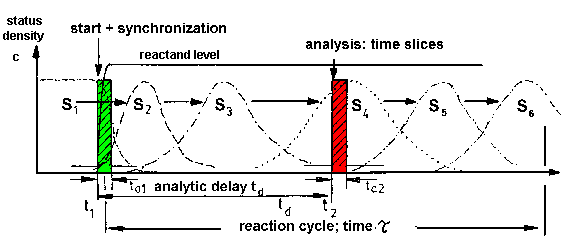 |
| Fig.1: a) The conversion of a chemical
or biological system leads from a start status (S) to a product status
(P) by a stepwise conversion at the single molecule level. In the
example a six status conversion is assumed, e.g. intermediate states of
an enzyme catalysis.
b) The investigation of a macroscopic sample (many molecules) has to be started by a jump event, e.g. by mixing of enzyme and substrate leading to substrate saturation (c >> Km). If this process has the speed control on the reaction, this leads to a synchronization of the sample for a limited time. The synchronized sample undergoes a transition of state densities S1 to S6 of molecules beeing in the status Z1 to Z6 in the example. The properties of the intermediates and their interconversions can be studied by time resolved investigation online, or after time resolved intermediate fixing in aliquots off-line (frozen intermediates). |
In some cases the process can be studied at the molecular level by single
molecule spectroscopy, e.g. with a molecule fixed in the beam of a confocal
laser microscope. This is difficult and risky due to radiation damage and
other artefact problems. For structural biology investigations those concepts
are planned for the free electron laser X-ray sources FEL at Stanford,
USA, and DESY, Hamburg (Ref.: Anfinrud
et al.; Nature 2000). In that particular case the undisturbed structure
is obtained by an ultrafast investigation in femtoseconds prior to radiation
demage (single molecule "crystallography without crystals").
In most other systems the conversion has to be studied at the macroscopic
level, i.e. using large samples containing millions of molecules,according
to Fig.1. This requires the synchronization of the reaction by a jump event,
e.g. by activating or adding substrate to an enzyme in a time shorter than
the duration of the reaction cycle; or by a temperature jump of a premix.
In light dependent systems this can be obtained by flash illumination.
The synchronized system can then be followed either: i) online by time
resolved investigation; or ii) off-line by static investigation after time
resolved intermediate fixation, e.g. by taking aliquots and rapid cooling
or freezing (fixed intermediates). Examples from the MPSD group are for
the online method (i) the time resolved structure
investigation of working F1ATPase after ATP-addition by TR-SAXS
at the ESRF, Grenoble, and the time
resolved study of lipid membrane structure after a pH-jump by time
resolved neutron TR-SANS scattering at the ILL,
Grenoble; and with the off-line method (ii) the time resolved investigation
of the light-driven ATP synthesis of co-reconstituted ATP-synthase/mBR
(Bacteriorhodopsin) liposomes as has been done e.g. in [F17,
F18].
(iii) As shown below we have recently developed a novel strategy, which
combines both methods in the rapid-mix/cold-trap method for the double
time resolved investigation of intermediates of very short live time in
enzymatic rection cycles.
Table.1: A process requires several start conditions. Only one or a few control speed and may thus be used for process synchronization.
|
|
|
| - have to be present for the process
- do not directly affect the rate limiting step |
- have to be present for the process
- affect the rate limiting step directly |
In any case of a study with a macroscopic sample, the success of the
time resolved investigation depends on the start and synchronization reaction.
According to table1, the start may depend on several conditions, which
are depicted as "necesseary conditions". Only one or a few lead to a synchronization
of the process, if they are controlling speed by direct access to the rate
limiting step.
If a reaction is to investigated by time resolved methods, the number
and nature of the start conditions has to be investigated carefully. If
both classes of conditions are present, the reaction has to be started
in a two-step process: i) the necessary conditions have to be established,
and ii) the speed controlling conditions have to be produced by a jump
event in a short time.
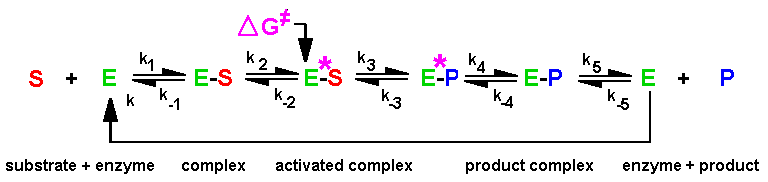 |
| Fig.2: The kinetics of most enzymes can be described by the Michaelis-Menten model. At low concentrations the speed is diffusion-controlled by the formation of the enzyme-substrate complex (ES) from free enzyme (E) and substrate (S). At high substrate concentration (c >> Km) most of the enzyme is present as ES complex. In that case the speed control is in the formation of the transition state ES*. The further reaction lead over the enzyme-product complexes (EP*, EP) to enzyme and product (P). Thus in many cases a substrate concentration jump cj > 2 Km leads to sample synchronization for a limited time. |
As depicted in Fig.2, in a simple enzyme catalysis the speed control
under substrate (S) saturation is according to the Michaelis-Menten kinetics
in the second reaction step, which leads from the enzyme-substrate complex
(ES) to the transition state ES* and then over the activated (EP*) and
relaxed enzyme-product comples (EP) to free enzye and product (P). If the
substrate (S) binding to the enzyme (E) is faster than the formation of
the transition state (ES*) from the enzyme-substrate complex (ES), the
reaction can be synchronized for one reaction cycle (at least ). According
to Equ.1 and this requires a concentration jump of the substrate to cj
> 2Km in a time much shorter than then duration of the reaction
cycle tr (Km is the Michaelis constant, i.e. the substrate
concentration where 50% of the enzyme in present as ES). In our experiments
with working F1ATPase and ATP-synthase
we use a substrate concentration jump (in ATP) to 8*Km in
0.2 - 5% of tr (depending on the temperature = speed). Note that
the nature of the rate control in a complex multidomain enzyme as ATPase
is not as simple as illustrated in Fig.2 !
|
|
cj > 2 Km |
|
|
tr = 1 / kcat |
Two examples may illustrate this important step in time resolved studies:
A) The study of the time coarse of subreactions
of working F1ATPase requires a preparation of pure, 100% reaction
competent and kinetically homogenous enzyme as necessary condition and
the rapid addition (stopped flow tchnique) or activation (caged-ATP flash
technique) of substrate (ATP) as speed controlling condition.
B) The study of reaction steps during ATP-synthesis of ATP-synthase/mBR
liposomes requires two pure and functional enzymes (ATP-synthase, monomeric
Bacteriorhodopsin mBR), liposomes and unique reconstitution as necessary
conditions. Furthermore the liposomes have to be "energized", i.e. the
generation of an electrochemical proton potential difference of the membrane
sufficient for ATP synthesis (delta-p > 100 mV). This is an important necesary
condition, but not the key for the rate limiting step ! The speed control
is at the nucleotide level as in (A), in this case by the substrate ADP.
Table.2: Techniques of active sample generation
| technique | time resolution | remarks |
| direct mixing | 1 s | eldest technique; mostly suitable at the system, but not at molecular event level |
| stopped flow rapid mixing | 1 ms | speed depends on supplied volume
additional delay (start to first data point) |
| photoactivation by a light pulse | fs | photosynthesis and photoreactions;
flash tubes: > 1µs laser: fs |
| photolytic activation by cleavage of a precursor, or by a radical (footprinting) | µs to ms; 25 ms for caged-ATP at pH7 | caged compounds are photo-cleaved in a 2-step process. Only the first is fast, the second (anion liberation, if any) depends on pH in speed. |
| photoexcitation of an electron system | fs to ns; < 5 ps for caged-acid proton | the secondary re-protonation of the slower anion has to be avoided for caged acids / proton |
| photolabeling and -crosslinking | µs | lifetime of active species (nitrene, carbene) has to be low (ns) for
structure specifity (epitope);
requires flash photolysis for reliable results |
| temperature jump | ms to s if acchieved directly
ns to µs possible, if done indirectly |
indirect method is slow (+,- direction)
ms temperature jump by stopped-flow mixing (+,-) fast temperature jump by flash or laser pulse absorption (+ direction only) |
| pressure jump | µs | energy absorption at constat volume;
piezo technique |
| field jump | ps | direct: electrodes or magnets;
light absorption (grazing incidence) |
| light pulse | fs | flash tubes for moderate speed (>1 µs)
laser methods for fast reactions |
According to table2, the active sample can be produced by several techniques,
which include a jump event of a speed controlling condition.
The direct mixing is limited to rather slow reactions (> 1s), if assuming
a conventional (photometer) cuvette equipped with a magnetic stirrer. A
further problem of this technique is the production of analytic aliquotes
of a batch sample because of pressure limits (> 1 bar, if suction technique
is used).
For the investigation of subprocesses in enzyme catalysis and polymer
reactions the stopped-flow rapid mixing technique is mostly spread (due
to early work of B. Chance and Holzwart; Nobel award for TR-studies).
Table.3: Components of a setup for time resolved invstigations
| component | stopped-flow rapid mixing;
quenched-flow multiple mixing; continuous flow |
flash photolysis (caged compounds);
photolabeling, photoactivation |
| sample supply unit | syringes for stock solutions on a common drive | syringe for premix solution on a drive |
| temperature conrol device | heat exchanger (umbilical unit) | heat exchanger (umbilical unit) |
| sample activation device | mixer; tangential flow or turbid stream | irradiation cuvette with two (UV)-light sources
mostly = sample investigation unit |
| sample investigation unit | pressure resistant cuvette, capillary | flat window cell, cuvette, capillary |
As depicted in table3 and Fig.3, a setup for time resolved investigations
consists of four devices at least: the sample supply unit, the temperature
conrol device, the sample activation device and the sample investigation
unit. Additionally tubing between the devices is required (PTFE tubing
or of coarse steel or PEEK capillaries, as usual in HPLC). The stopped-flow
technique is expensive and limited to moderate speed (>1 ms), but rather
easy to use. The flash photolysis is easy in principle and does not include
an unintended pressure jump or shearing forces inside the sample. Thus
a flat window cell can be used for the in situ activation
and investigation of the sample. The problem is here the more complicated
chemistry and the unique distribution of light inside the sample. For both
problems solutions are presented in the optics
chapter and the chemical synthesis page.
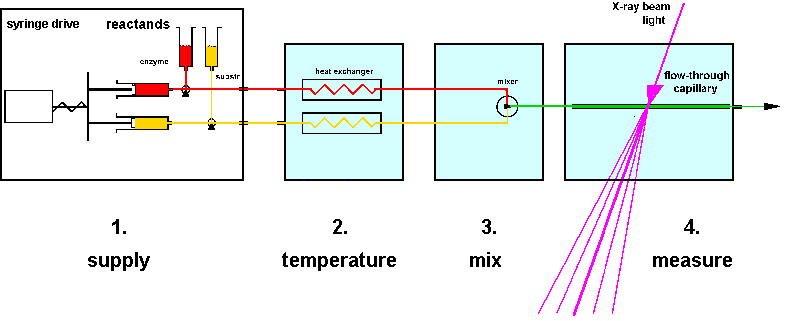 |
| Fig.3: A setup for time resolved investigations consists of four components at least: 1) the sample delivery unit; 2) the temperatur control device; 3) the sample activation device (mixer, flash unit); and 4) the sample investigation unit. Two or more components may be combined to a compact-device. |
The four units of a timer resolved setup may be combined to a compact block, as is the case with many commercial stopped-flow devices (e.g. from Biologique, Grenoble), or they may be constructed as separate devices, as is the case with several commercial components (e.g. SF20mXM from HighTech Scientific, UK) and in our own constructions (stopped-flow devices MSF1 to MSF4 and sample environments, in part with helium-jet cooling and additional temperature jump / cold trap unit). As shown in table4, each solution has advantages and disadvantages, depending on the application:
Table.4: Advantages of compact-device and separate-component setups for time resolved investigations.
| concept | advantage | disadvantage |
| compact-device | easy to use
fast setup low dead volume |
low flexibility in sample conditions
time-consuming sample change and flush only one temperature possible |
| separate-component | high flexibility of conditions
additional methods possible, e.g. dual beam, UV-flash, light guides, sample camera control etc. temperature jump possible (cold trap method) helium-cooling with separate sample box, low background with helium atmosphere |
complicated setup
slow setup (hours) dead volume in connecting tubings (some 100 µl) voluminous equipment weight (SBox4 with Alu-Lead shielding = 30 kg) |
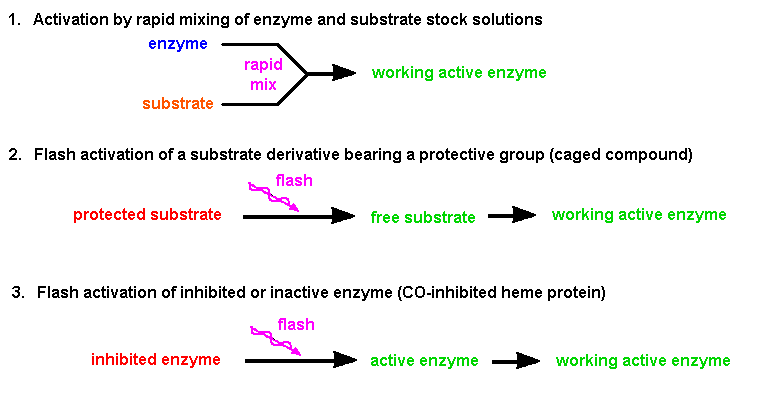 |
| Fig.4: The time resolved investigation of a running process requires the activation in a short time. This trigger can be achieved in three ways: i) by rapid mixing (fluids), ii) by flash activation of the substrate, or iii) by flash activation of an inhibited enzyme (from T20). |
The online study of a running process is the most direct access to conversions and intermediates. In case of rapid reactions, which only scarcely depend on temperature, it is the only direct experimental method, due to the limited time resolution of available methods for intermediate fixation, e.g. using stopped-flow devices (tr > 1 ms). Technically the online observation can be done in tthree ways by: i) continuous flow investigation, or ii) stopped-flow experiments, which are for structure research done in a favorate way as multiple mixing quenched flow variant with a stepper motor driven multiple shot stopped-flow device; iii) A third technique is the observation of the process after in-situ activation by a light flash, e.g. photolysis of a caged compound, photoactivation of an enzyme or substrate, or other photoreactions. The latter method applies to crystallography with X-ray and neutron radiation and to the investigation of sensitive samples by scattering and spectroscopy.
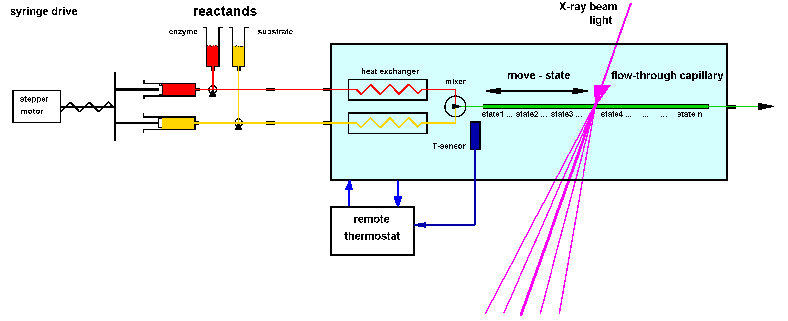 |
| Fig.5: The time resolved investigation of a running process using the continous flow technique requires a significant sample volume. During the whole experiment active sample is produced continuously in the mixer and flows through the long capillary, which is scanned by the experiment, e.g. a X-ray beam. The distance between mixer and test point correlates with the reaction time und thus the molecular status. |
The principle of an continous flow experiment is depicted in Fig.4.
This type of simple time resolved experiment is favorate, if a large amount
of the sample is available, e.g. during the investigation of chemical reaction
(polymerization). In structural biology mostly a few milliliters of protein
solution containing typically 1% protein or less is available. Thus the
stopped-flow technique or flash photochemistry methods using a discontinous
flow are applied here. Examples of those setups for time resolved X-ray
small angle scattering TR-SAXS and time resolved neutron small angle scattering
TR-SANS which have been develloped by the MPSD group are shown in Fig.6
und Fig.7. Both setups use the multiple shot quenched flow variant with
stepping motor stopped-flow devices. This allows several shots without
breaking the radiation security interlock system. The setups were used
for the investigation of F1ATPase, ATP-Synthase, GroEL, Myoglobin
and liposomes in cooperation with H. Heumann and M. Rössle, MPI Martinsried;
W. Doster, TU München; T. Narayanan, O. Diat and P. Boesecke, ESRF
Grenoble; H. Amenitsch and S. Bernstorff, ELETTRA Trieste; and R.P. May
and J. Holzinger, ILL Grenoble.
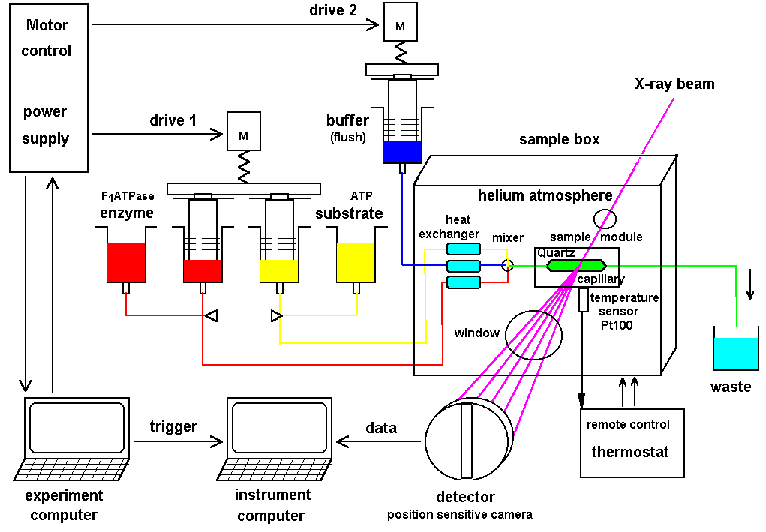 |
| Fig.6: Setup for the investigation of a working enzyme by time resolved X-ray small angle scattering TR-SAXS. The active sample is produced after thermalisation and mixing inside the external sample environment box (SBox1, 2). The helium atmosphere reduces the background of air-scattering and allows definite temperature control. SBox2 and 4 are equipped with an addition helium-jet cooling of the sample irradiation point to avoid beam heating. By the second drive the sample can be slowly replaced be fresh working solution from the loop before the cell by drag buffer (flux: 0.1 - 5 µl/s) [from R6]. |
The X-ray setup (Fig.6) uses the sample cell in a flow-through mode.
about 50% of the new shot is here used for removal of the old sample and
flushing. The working solution between mixer and irradiation point (80
µl in our helium filled sample environments
SBox1, 2 and 4) can be used for slow continuous sample replacement
during the exposure in order to avoid radiation demage artefacts (drag
buffer, flux = 0.1 to 2 µl/s). The setup was used as depicted at
the ESRF beamlines ID2A and ID24 and at ELETTRA beamline SAXS (5.2); the
helium-jet
cooled sample environment SBox4 (without stopped-flow) was used at
ESRF-ID1 and DESY-HASYLAB beamline USAXS (BW4).
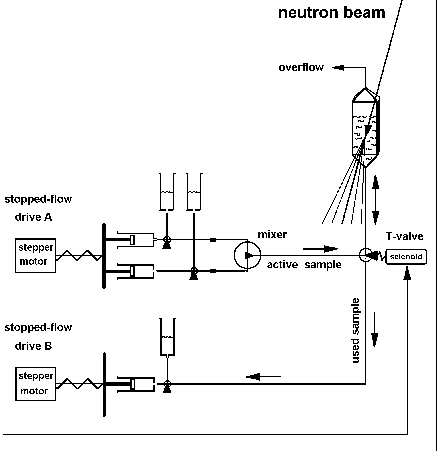 |
Fig.7: Setup for time resolved neutron small angle scattering TR-SANS. The active sample is produced in the external mixer from stock solutions and transfered via a 3-way valve into the quartz cuvette, which is filled only by 2/3. After the electronic exposure of the structural film the T-valve is switched over and the sample is removed backwards using the second drive. The upper connection is an air and emergency outlet [from T20, R10]. |
In the TR-SANS setup (Fig.7) the cuvette (d= 1 mm or 10 mm; Hellma)
is only filled by 2/3. The upper outlet is an emergency and air exit. After
the TR-exposure the T-valve between mixer and cuvette is switched over
and the sample is removed with the second drive, which moves the waste
syringe. The setup was developped for the time resolved investigation of
liposomes and GroEL at the ILL beamline D22 (see preliminary reports R10,
R11).
Technical details of our sample environments and our mixing devices
are shown in at the rapid mixing technology page.
In
situ activation of crystals, solids and sensitive samples
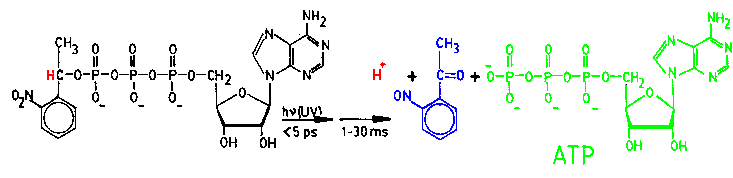 |
| Fig.8: The flash photolysis of a caged compound generates a concentration jump of the fragments, e.g. ATP and protons [from T20]. |
During the time resolved investigation by crystallography with X-rays
and neutrons and by solution scattering of samples sensitive to shearing
forces the process by activation mixing does cannot be applied. Thus a
substrate concentration jump required as trigger of the process has to
be generated in-situ. As depicted in Fig.8, this can be done by flash phtolysis
of compounds bearing a photo-lable protective group, e.g. a caged compound.
The most simple caged compounds are esters of 2-nitro-phenyl-ethanol and
an acid, e.g. ATP. Those substrates, e.g. caged-ATP, may bind to the catalytic
center of an enzyme or not, depending on the protein. Both classes of enzymes
may be investigated with a caged compound, as even a crystal contains enough
solvent to bear the caged substrate close to the enzyme (typically 70%
of the crystal mass is solvent). As shown at the chemical
synthesis page, the MPSD group synthesizes caged-nucleotides as well
as the novel caged-acids, which can generate a stable large pH-jump immediately
after flash photolysis (delta-pH > 2; dt ~ ps).
An alternative to the caged compound technology is the activation of
the protein by a flash. Several Heme-iron proteins can be inhibited with
carbon monoxide CO. Due to spectral changes by CO-inhibition, this inhibitor
can be eliminated by a light flash (femtoseconds). This technique has been
used for the investigation of cytochrome-oxidases in Cooperation with G.
Heinz and W. Doster (TU Munich) (see T21).
A sketch of typical CO-inhibition and flash re-activation sequence is depicted
in Fig.9.
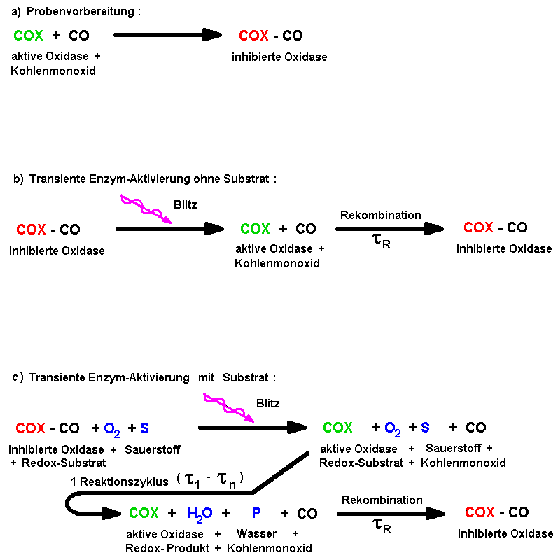 |
| Fig.9: Heme-iron proteins as Cytochrom-c oxidase can be inhibited by carbon monoxide (a) and reactivated by a light flash (b). After flash photolysis in presence of substrate (c), a single enzymatic reaction cycle occurs, because the recombination of the inhibitor is slower than the catalysis. |